

CUT&RUN Assay Kit (with Drosophila Spike-In Control) のプロトコール
| ! | この!マークは、実施する CUT&RUN反応数に応じて量を変更する、プロトコール中の 重要なステップであることを意味します。 |
| !! | この!!マークは、操作を進める前に バッファー を希釈する、重要なステップであることを意味します。 |
| SAFE STOP | これは、実験操作を中断する必要がある場合に、プロトコールを安全に中断できるポイントを示します。 |
I. Concanavalin Aビーズの活性化
実験開始前の準備:
!すべてのバッファーの量は、実施するCUT&RUN反応数に応じて比例的に増加させる必要があります。
Concanavalin A Bead Activation Bufferを氷上で保持してください。
- 実行するCUT&RUN反応の数を決定してください。ポジティブコントロールのTri-Methyl-Histone H3 (Lys4) (C42D8) Rabbit Monoclonal Antibody #9751およびネガティブコントロールのRabbit (DA1E) Monoclonal Antibody IgG Isotype Control(CUT&RUN) #66362を反応を含めることを強く推奨します。
- Concanavalin A磁気ビーズを、ビーズ懸濁液がチューブからこぼれないように慎重にピペッティングで上下させて再懸濁してください。
- CUT&RUN 1反応につきビーズ懸濁液10 µLを、新しい1.5 mLチューブに移してください。一度に14回を超えるCUT&RUN反応を行う場合は、2本以上の1.5 mLチューブを使用してください。各1.5 mLチューブに140 µL以上のConcanavalin Aビーズを入れないでください。
- ビーズ10 µLにつき、Concanavalin A Bead Activation Buffer 100 µLを加えてください。ピペッティングで穏やかに上下させてビーズを混合してください。
- チューブを溶液が透明になるまで磁気ラックに置き (30秒間から2分間)、その後、液体を取り除き廃棄してください。
- チューブを磁気ラックから外してください。ステップ4-5をもう一度繰り返して、2回目のビーズの洗浄を実施してください。
- ビーズ懸濁液の最初の量と同量 (1サンプルにつき10 µL) のConcanavalin A Bead Activation Bufferを加えて、ピペッティングで上下させて再懸濁してください。 注意:活性化したビーズは氷上で最長8時間保管できます。
注意:ボルテックスを繰り返すとコンカナバリンAがビーズから外れてしまう場合があります。Concanavalin A磁気ビーズをボルテックスしないでください。
注意:ビーズの損失を避けるため、プロトコールのいずれの時点においても真空吸引はしないでください。
II. 細胞と組織のサンプルの調製
ほぼ全ての細胞タイプにおいて、生細胞を用いたCUT&RUNアッセイにより、ヒストンや転写因子、コファクターを安定して濃縮できます。Concanavalin Aにより損傷を受ける、または感受性のある細胞の場合は、軽く固定することにより細胞を無傷のまま保存できます。また、新鮮な細胞を用いても安定したシグナルがみられない場合に、軽く固定を行うことにより、存在量の少ない、あるいは結合力の弱い転写因子やコファクターの濃縮を促進できる場合があります。細胞の過剰な固定はCUT&RUNアッセイを阻害する可能性があることに注意してください。
弊社のCUT&RUNアッセイで、広範囲な細胞や組織サンプルを解析できます。プロトコールに記載されているように、1回あたり5,000-250,000個の細胞または1-5 mgの組織を用いてCUT&RUN反応を行うことができます。この範囲であれば、プロトコール全体を通して使用するバッファーの量を1反応あたりの細胞や組織の量に応じて調整する必要はありません。指示がある場合には、実施する反応数に応じてバッファーの量を比例的に増やす必要があります。可能であれば、1反応につき100,000個の細胞または1 mgの組織を使用することを推奨します。細胞数に限りがある場合、ヒストン修飾の解析には1反応につき少なくとも5,000-10,000個の細胞を、転写因子やコファクターの解析には1反応につき10,000-20,000個の細胞を使用することを推奨します。
A. 生細胞サンプルの調製
実験開始前の準備:
!すべてのバッファーの量は、実施するCUT&RUN反応数に応じて比例的に増加させる必要があります。
Protease Inhibitor Cocktail (200X) #7012 (PIC) と100X Spermidine #27287を温めてください。どちらも必ず完全に解凍してください。Protease Inhibitor Cocktail (200X) #7012はDMSO含有のため、氷上に置くと再凍結することにご注意ください。
1X Wash Buffer (1細胞株あたり2 mL、1反応あるいは1インプットサンプルにつき追加で100 µL) を調製してください。例えば、1X Wash Buffer 2.5 mLを調製する場合は、10X Wash Buffer (CUT&RUN, CUT&Tag) #31415 250 µLに100X Spermidine #27287 25 µLとProtease Inhibitor Cocktail (200X) #7012 12.5 µL 、Nuclease-free Water #12931 2,212.5 µLを加えてください。細胞へのストレスを最小限にするため、室温に戻してください。
注意:生細胞 (未固定) サンプル調製のステップは、細胞へのストレスを最小限にするため、室温で連続して行う必要があります。DNAの断片化を最小限にするため、再懸濁する場合には激しいボルテックスや気泡の混入を避けてください。
- 細胞ストレスを最小限にするため、新たに培養した細胞を室温で回収してください。各反応あたり5,000-100,000個の細胞を回収し、さらにインプットサンプルの調製用に5,000-100,000個の細胞を回収してください。必ず、ポジティブコントロールのTri-Methyl-Histone H3 (Lys4) (C42D8) Rabbit Monoclonal Antibody #9751およびネガティブコントロールのRabbit (DA1E) Monoclonal Antibody IgG Isotype Control) (CUT&RUN) #66362を反応数に含めてください。
- 細胞懸濁液を600 x gで3分間、室温で遠心分離して、液体を除去し廃棄してください。
- 1X Wash Buffer (+ Spermidine + PIC) 1 mLを室温で加え、ピペッティングで穏やかに上下させて細胞ペレットを再懸濁してください。
- 600 x gで3分間、室温で遠心分離し、液体を除去して廃棄してください。
- ステップ3-4を繰り返して、もう1度細胞ペレットを洗浄してください。
- 各反応またはインプットサンプルあたり、1X Wash Buffer (+ Spermidine + PIC) 100 µLを加え、ピペッティングで静かに上下させて細胞ペレットを再懸濁してください。
- 細胞100 µLを新しいチューブに移して、セクションVIIを実施するまで4°Cで保管してください。これがインプットサンプルです。
- 速やかにセクションIIIに進んでください。
注意:接着細胞を回収する場合は、トリプシン処理で細胞を培養ディッシュから剥離させた後、3倍量以上の組織培養液で反応を停止させてください。スクレイパーを用いて培養皿から細胞を掻き取ると、ストレスの原因となり細胞を溶解させてしまう場合もあるため、CSTはこれを推奨しません。適切な数の細胞を実験に用いるため、血球計算盤やその他の細胞計数装置で細胞数をカウントしてください。
注意:総細胞数が100,000個未満で、遠心分離した細胞ペレットが眼で見えない場合は、洗浄ステップ中に細胞を損失しやすくなります。したがって、少数の細胞で実験を進める場合は下記ステップ3-5の洗浄ステップの省略を推奨します。懸濁液に40%の細胞培地が混入していても Concanavalin Aビーズと細胞は結合します。そのため、ステップ2の細胞懸濁液の初めの遠心分離の後に、上清の大部分を除去して廃棄し、1反応あたり≤40 µLの細胞培地を残すことができます。この場合、ステップ6で細胞懸濁液に十分量の1X Wash Buffer (+ Spermidine + PIC) を加え、1反応あたりの合計容量を100 µLにしてください。
注意:このインプットサンプルは、後のプロトコールで55°Cでインキュベートするので、インキュベート中の蒸発を減少させるためセーフロックの付いた1.5 mLチューブを使用することを推奨します。
B. 固定細胞サンプルの調製
注意:固定細胞サンプルの調製には、本キットに含まれない次の試薬が必要です:37% Formaldehydeまたは16% Formaldehyde Methanol-Free #12606、Glycine Solution (10X) #7005、10% SDS Solution #20533
実験開始前の準備:
!すべてのバッファーの量は、実施するCUT&RUN反応数に応じて比例的に増加させる必要があります。
Protease Inhibitor Cocktail (200X) #7012と100X Spermidine #27287を温めてください。どちらも必ず完全に解凍してください。Protease Inhibitor Cocktail (200X) #7012はDMSO含有のため、氷上に置くと再凍結することにご注意ください。
1X Wash Buffer (1細胞株につき2 mL、1反応あるいは1インプットサンプルにつき追加で100 µL) を調製してください。例えば、1X Wash Buffer 2.5 mLを調製する場合は、10X Wash Buffer (CUT&RUN, CUT&Tag) #31415 250 µLに100X Spermidine #27287 25 µLとProtease Inhibitor Cocktail (200X) #7012 12.5 µL 、Nuclease-free Water #12931 2,212.5 µLを加えてください。細胞へのストレスを最小限にするため、室温に戻してください。
固定処理を行う細胞懸濁液1 mLにつき、37% Formaldehydeを2.7 µLまたは16% Formaldehyde Methanol-Free #12606を6.25 µL取り分け、室温で保持してください。メーカーが示す使用期限内の新しいホルムアルデヒドを使用してください。
- 各反応あたり5,000-100,000個の細胞を回収し、さらにインプットサンプルの調製用に5,000-100,000個の細胞を回収してください。必ず、ポジティブコントロールのTri-Methyl-Histone H3 (Lys4) (C42D8) Rabbit Monoclonal Antibody #9751およびネガティブコントロールのRabbit (DA1E) Monoclonal Antibody IgG Isotype Control) (CUT&RUN) #66362を反応数に含めてください。
- 細胞懸濁液1 mLにつき、37% Formaldehydeを2.7 µLまたは16% Formaldehyde Methanol-Free #12606を6.25 µL加え、ホルムアルデヒドの最終濃度が0.1%となるように調整してください。チューブを転倒混和しながら室温で2分間インキュベートしてください。
- 固定処理した細胞懸濁液1 mLあたり、Glycine Solution (10X) #7005を100 µL加え、クロスリンクを停止してください。チューブを転倒混和しながら室温で5分間インキュベートしてください。
- 細胞懸濁液を3,000 x gで3分間、4°Cで遠心分離して、液体を除去し廃棄してください。速やかにステップ5に進んでください。(SAFE STOP) 固定した細胞ペレットは、使用するまで-80°Cで最長6か月間保存できます。
- 1X Wash Buffer (+ Spermidine + PIC) 1 mLを加え、ピペッティングで穏やかに上下させて細胞ペレットを再懸濁してください。
- 3,000 x gで3分間、4°Cで遠心分離して、液体を除去し廃棄してください。
- ステップ5-6を繰り返して、もう1度細胞ペレットを洗浄してください。
- 各反応またはインプットサンプルあたり、1X Wash Buffer (+ Spermidine + PIC) 100 µLを加え、ピペッティングで静かに上下させて細胞ペレットを再懸濁してください。
- 細胞100 µLを新しいチューブに移して、セクションVIIを実施するまで4°Cで保管してください。これがインプットサンプルです。
- 速やかにセクションIIIに進んでください。
注意:接着細胞株を回収する場合は、トリプシン処理で細胞を培養ディッシュから剥離させた後、3倍量以上の組織培養液で反応を停止させてください。スクレイパーを用いて培養皿から細胞を掻き取ると、ストレスの原因となり細胞を溶解させてしまう場合もあるため、CSTはこれを推奨しません。適切な数の細胞を実験に用いるため、血球計算盤やその他の細胞計数装置で細胞数をカウントしてください。
注意:総細胞数が100,000個未満で、遠心分離した細胞ペレットが眼で見えない場合は、洗浄ステップ中に細胞を損失しやすくなります。この場合は細胞ペレットの凍結保存を推奨しません。このような少数の細胞で実験を進める場合は下記ステップ5-7の洗浄操作を省略することを推奨します。懸濁液に40%の細胞培地が混入していても Concanavalin Aビーズと細胞は結合します。そのため、ステップ4の細胞懸濁液の初めの遠心分離の後に、上清の大部分を除去して廃棄し、1反応あたり≤40 µLの細胞培地を残すことができます。この場合、ステップ8で細胞懸濁液に十分量の1X Wash Buffer (+ Spermidine + PIC) を加え、1反応あたりの合計容量を100 µLにしてください。
注意:このインプットサンプルは、後のプロトコールで55°Cでインキュベートするので、インキュベート中の蒸発を減少させるためセーフロックの付いた1.5 mLチューブを使用することを推奨します。
C.組織サンプルの調製
ほぼ全ての組織タイプにおいて、軽く固定した (0.1%ホルムアルデヒドで2分間) 組織1 mgを用いることにより、ヒストンや転写因子、コファクターを安定して濃縮できます。ヒストン修飾の濃縮の場合は、ホルムアルデヒド固定は必須ではありません。しかし、多くの転写因子やコファクターの場合、軽く固定することで最適な結果が得られます。存在量の少ない、あるいは結合力の弱い転写因子やコファクターの場合は、中程度の固定 (0.1%ホルムアルデヒドで10分間) が必要なこともあります。また、線維組織のような難しい組織を使用する場合には、中程度の固定によって結果が改善されることがあります。過剰な固定はCUT&RUNアッセイを阻害する可能性があることに注意してください。固定した組織サンプルは、凍結することにより使用するまで-80°Cで最長6か月間保存できます。
注意:固定組織サンプルの調製には、本キットに含まれない次の試薬が必要です:37%ホルムアルデヒドまたは16% Formaldehyde Methanol-Free #12606、Phosphate Buffered Saline (PBS-1X) pH7.2 (Sterile) #9872、Glycine Solution (10X) #7005、および10% SDS Solution #20533
実験開始前の準備:
!すべてのバッファーの量は、実施するCUT&RUN反応数に応じて比例的に増加させる必要があります。
Protease Inhibitor Cocktail (200X) #7012と100X Spermidine #27287を温めてください。どちらも必ず完全に解凍してください。Protease Inhibitor Cocktail (200X) #7012はDMSO含有のため、氷上に置くと再凍結することにご注意ください。
- 1X Wash Buffer (1組織タイプにつき3 mL、1反応あるいは1インプットサンプルにつき追加で100 µL) を調製してください。例えば、1X Wash Buffer 3.5 mLを調製する場合は、10X Wash Buffer (CUT&RUN, CUT&Tag) #31415 350 µLに100X Spermidine #27287 35 µL、Protease Inhibitor Cocktail (200X) #7012 17.5 µL、Nuclease-free Water #12931 3,097.5 µLを加えてください。細胞へのストレスを最小限にするため、室温に戻してください。
組織の固定が必要な場合は次のバッファーを調製してください:
- 1組織につき1 mLの固定バッファーが必要です。Phosphate Buffered Saline (PBS-1X) pH7.2 (Sterile) #9872 1 mLに、37% formaldehyde 2.7 µLまたは16% Formaldehyde, Methanol-Free #12606 6.25 µL、Protease Inhibitor Cocktail (200X) #7012 5 µLを加えて調製してください。メーカーが示す使用期限内の新しいホルムアルデヒドを使用してください。
- 各組織の種類ごとに、Phosphate Buffered Saline (PBS-1X) pH7.2 (Sterile) #9872 1 mL + Protease Inhibitor Cocktail (200X) #7012 5 µLを調製し、氷上で保持してください。
- 固定バッファー1 mLにつき、Glycine Solution (10X) #7005 100 µLを調製してください。
- 1抗体反応につき新鮮な組織を1 mg測り取り、さらにインプットサンプル調製用に1 mgを測り取ってください。必ず、ポジティブコントロールのTri-Methyl-Histone H3 (Lys4) (C42D8) Rabbit Monoclonal Antibody #9751およびネガティブコントロールのRabbit (DA1E) Monoclonal Antibody IgG Isotype Control) (CUT&RUN) #66362を反応数に含めてください。
- 組織サンプルをシャーレに置き、清潔な解剖用メスまたは剃刀の刃で細かく切り刻んでください。ディッシュは氷上で保持してください。タンパク質分解を防ぐため、組織をよく冷やしてください。
- 刻んだ組織を速やかにFixation Buffer 1 mLに移し、チューブを転倒混和してください。
- 室温で2分間インキュベートしてください。
- 固定バッファー1 mLにつき、Glycine Solution (10X) #7005 100 µLを加えてクロスリンクを停止してください。チューブを転倒混和しながら室温で5分間インキュベートしてください。
- 2,000 x gで5分間、4°Cで組織を遠心分離し、液体を除去して廃棄してください。
- 1X PBS+PIC 1 mLに組織を再懸濁してください。
- 2,000 x gで5分間、4°Cで遠心分離し、液体を除去して廃棄してください。その後、ステップ9に進んでください。(SAFE STOP) 固定した組織ペレットは、解離する前に-80°Cで最長6か月まで保存できます。
- 組織を1X Wash Buffer (+ Spermidine + PIC) 1 mLで再懸濁し、サンプルをDounce Homogenizerに移してください。
- 組織片を、シングルセル懸濁液になるまで破砕してください。20-25ストロークで、組織の塊が見られなくなるまで破砕してください。
- 細胞懸濁液を1.5 mLチューブに移し、3,000 x gで3分間、室温で遠心分離し、細胞から上清を除去して廃棄してください。
- 細胞ペレットを1X Wash Buffer (+ Spermidine + PIC) 1 mLで再懸濁してください。
- 細胞懸濁液を3,000 x gで3分間、室温で遠心分離し、液体を除去して廃棄してください。
- ステップ12-13を繰り返して、もう1度細胞ペレットを洗浄してください。
- 1反応につき、1X Wash Buffer (+ Spermidine + PIC) 100 µLを加え、ピペッティングで穏やかに上下させて細胞ペレットを再懸濁してください。
- 細胞100 µLを新しいチューブに移して、セクションVIIを実施するまで4°Cで保管してください。これがインプットサンプルです。
- 速やかにセクションIIIに進んでください。
注意:転写因子やコファクターを解析する場合や、線維組織のような難しい組織を使用する場合は、1反応あたり5 mgまでの組織を試薬量を増やすことなく使用できます。
注意:ほとんどの組織タイプや標的タンパク質は、軽く固定することで最適な結果が得られます。このため、次の軽く固定するプロセスの実施を推奨します。しかし、新鮮組織 (未固定) が望ましい場合は、ステップ3-8を省略して直ちにステップ9に進んでください。
注意:この量の固定バッファーで、最大50 mgの組織を固定処理できます。>50 mgの組織を処理する場合は、固定バッファーとステップ7の1X PBS+PIC液をスケールアップしてください。
注意:線維組織などの難しい組織を使用する場合や、存在量の少ないあるいは結合力の弱い転写因子やコファクターを解析する場合は、ホルムアルデヒドによる固定を10分間に延長することで結果が改善される場合があります。
注意:このインプットサンプルは、後のプロトコールで55°Cでインキュベートするので、インキュベート中の蒸発を減少させるためセーフロックの付いた1.5 mLチューブを使用することを推奨します。
III. サンプル正規化のためのDrosophila Spike-In Nuclei Controlの添加
実験開始前の準備:
Drosophila Spike-In Nuclei Controlを室温で解凍してください。1反応につき核Spike-in 20 µLを用いるという推奨条件に基づき、各製品バイアルには100,000-250,000個の細胞または1-2.5 mgの組織を含む反応を最大8回行うのに十分な量の核Spike-inが入っています。
1回の実験で2バイアル以上のDrosophila Spike-In Nuclei Controlが必要な場合は、各反応に添加する核Spike-in数の一貫性を確保するため、解凍済みバイアルをプールしてください。
Drosophila Spike-In Nuclei Controlの凍結および解凍は最小限に抑えてください。凍結融解回数を4回以下に抑えるため、より小さな容量に分注してください。この回数を超えると、Drosophilaゲノムへのマッピング率が低下し、1反応あたりに必要な核Spike-in量が増える可能性があります。例えば、9回の凍結解凍サイクルでは、Drosophilaへのマッピング率が約50%低下する可能性があります。
- 1反応につき100 µLのセクションII-Aのステップ6、セクションII-Bのステップ8、またはセクションII-Cのステップ15で調製した細胞懸濁液を1.5 mLチューブに分注してください。
- 1反応 (100,000-250,000個の細胞または1-2.5 mgの組織) につき、Drosophila Spike-In Nuclei Controlを20 µL加えてください。細胞と核Spike-inのこの比率により、通常、DrosophilaのSpike-in正規化リードが全シーケンシングリードの約0.5-10%を占める結果が得られます。
- 100,000個未満の細胞または1 mg未満の組織を使用する場合は、Drosophila Spike-In Nuclei Controlの容量を比例的に減らし、100,000細胞あたりまたは1 mgの組織あたり20 µLの比率を維持してください (例:50,000個の細胞に対しては10 µL、5,000個の細胞に対しては1 µL)。
- また、標的分子が低発現 (例:特定の転写因子またはコファクター) の場合は、核Spike-inの量を2-10分の1に減らす必要があるかもしれません。例えば、1反応 (100,000-250,000個の細胞または1-2.5 mgの組織を含む) につき、Drosophila Spike-In Nuclei Controlを2-10 µL加えてください。
- Drosophila Spike-In Nuclei Controlを含むCUT&RUN反応は、下流のqPCR解析および次世代シーケンシング (NGS) 解析の両方を正規化できます。いずれのアッセイも同量の核Spike-inを使用するため、反応を2回分用意する必要はありません。
- 穏やかにピペッティングで上下させて混合してください。
- 速やかにセクションIVに進んでください。
注意:CUT&RUNアッセイにおけるDrosophila Spike-In Nuclei Controlの至適用量は一定ではなく、Spike-in正規化リードが全シーケンシングリードの約0.5-10%となるように、お客様ご自身で最適化する必要があります。
IV. Concanavalin Aビーズと一次抗体の結合
実験開始前の準備:
注意:細胞の透過化に用いるジギトニンの推奨量は過剰量であり、ほとんどの細胞株や組織を十分に透過化できます。ただし、すべての細胞株や組織が、ジギトニンに対して同じ感受性を示すわけではありません。特定の細胞株や組織で推奨濃度のジギトニンが機能しない場合は、Appendix Aのプロトコールに従って条件を最適化してください。ジギトニン処理により、細胞集団の>90%が透過化される必要があります。
注意:セクションIV-VIの全インキュベーションステップにおいて、または回転によりサンプルを混合する必要はありません。チューブを指定の温度でラック上に静置するだけで構いません。混合してもアッセイ性能は向上せず、むしろビーズがチューブ壁やキャップに付着し、ビーズの凝集や損失につながる可能性があります。
!すべてのバッファーの量は、実施するCUT&RUN反応数に応じて比例的に増加させる必要があります。
Digitonin Solution #16359を90-100°Cで5分間温め、完全に解凍されて溶解していることを確認してください。解凍したDigitonin Solution #16359はすぐに氷上に置いてください。
Protease Inhibitor Cocktail (200X) #7012と100X Spermidine #27287を温めてください。どちらも必ず完全に解凍してください。Protease Inhibitor Cocktail (200X) #7012はDMSO含有のため、氷上に置くと再凍結することにご注意ください。
1反応につき、100X Spermidine #27287 1 µL + Protease Inhibitor Cocktail (200X) #7012 0.5 µL + Digitonin Solution #16359 2.5 µL + Antibody Binding Buffer (CUT&RUN, CUT&Tag) #15338 96 µLを調製し、氷上に置いてください (1反応あたり100 µL)。
注意:Digitonin Solution #16359は-20°Cで保管する必要があります。使用中は氷上に置き、その日の使用が終了したら-20°Cで保管してください。
- セクションIのステップ7で調製済みの活性化したConcanavalin Aビーズを室温にし、ピペッティングで穏やかに上下させて十分混合してください。
- 各チューブの細胞 + セクションIIIのステップ3で調製したDrosophila Spike-In Nuclei Control懸濁液に、10 µLのビーズ懸濁液を加えてください。
- ピペッティングで上下させてサンプルを十分に混合してください。室温で5分間インキュベートしてください。
- チューブを溶液が透明になるまで磁気ラックに置き (30秒間から2分間)、その後、液体を除去して廃棄してください。
- チューブを磁気ラックから外してください。Antibody Binding Buffer (+ Spermidine + PIC + Digitonin) 100 µLを各チューブに加え、ピペッティングで上下させて穏やかに混合し、氷上に置いてください。
- 各反応チューブに適量の試験抗体と1 µLのH2Av Rabbit Monoclonal Antibodyを加えて、ピペッティングで上下させて穏やかに混合してください。
- サンプルを4°Cで2時間、または室温で1時間インキュベートしてください。このステップは4°Cで一晩まで延長できます。
注意:CUT&RUN反応に必要な試験抗体の量は異なるため、お客様自身で決定してください。ポジティブコントロールのTri-Methyl-Histone H3 (Lys4) (C42D8) Rabbit Monoclonal Antibody #9751の場合は、サンプルに抗体2 µLを加えてください。ネガティブコントロールのRabbit (DA1E) Monoclonal Antibody IgG Isotype Control (CUT&RUN) #66362の場合は、サンプルに抗体5 µLを加えてください。「抗体なし」のコントロールの場合は、MNaseによる非特異的な消化とバックグラウンドノイズが高レベルになるため、ネガティブコントロール抗体を使用することを強く推奨します。qPCR解析とNGS解析の両方で比較するために、可能な限りインプットサンプルを用いることを推奨します。
V. pAG-MNase Enzymeの結合
実験開始前の準備:
!すべてのバッファーの量は、実施するCUT&RUN反応数に応じて比例的に増加させる必要があります。
Digitonin Solution #16359を90-100°Cで5分間温め、完全に解凍されて溶解していることを確認してください。解凍したDigitonin Solution #16359はすぐに氷上に置いてください。
Protease Inhibitor Cocktail (200X) #7012と100X Spermidine #27287を温めてください。どちらも必ず完全に解凍してください。Protease Inhibitor Cocktail (200X) #7012はDMSO含有のため、氷上に置くと再凍結することにご注意ください。
1反応につき、Digitonin Buffer [10X Wash Buffer (CUT&RUN, CUT&Tag) #31415 320 µL + 100X Spermidine #27287 32 µL + Protease Inhibitor Cocktail (200X) #7012 16 µL + Digitonin Solution #16359 80 µL + Nuclease-free Water #12931 2.752 mL] を3.2 mL調製してください。
新しいチューブに、1反応につきDigitonin Buffer (上記) 50 µLとpAG-MNase Enzyme 1.5 µLを加え、pAG-MNaseプレミックスを作製してください。例えば反応10回分の場合は、Digitonin Buffer 500 µLを新しいチューブに移し、pAG-MNase Enzyme 15 µLを加えてください。ピペッティングで上下させて混合し、氷上に置いてください。
注意:Digitonin Solution #16359は-20°Cで保管する必要があります。使用中は氷上に置き、その日の使用が終了したら-20°Cで保管してください。
注意:ここで調製したDigitonin BufferをセクションVとVIの両方で使用します。
- セクションIVステップ7のチューブを溶液が透明になるまで (30秒間から2分間) 磁気ラックに置き、その後、液体を除去して廃棄してください。
- チューブを磁気ラックから外し、Digitonin Buffer (+ Spermidine + PIC + Digitonin) 1 mLを加えてください。チューブの壁に張り付いたビーズを確実に回収できるように、穏やかに上下にピペッティングしてビーズを再懸濁してください。
- チューブを溶液が透明になるまで磁気ラックに置き (30秒間から2分間)、その後、液体を除去して廃棄してください。
- チューブを磁気ラックから外してください。各チューブにpAG-MNaseプレミックス50 µLを加えて、ピペッティングで穏やかに上下させてサンプルを混合してください。
- チューブを4°Cで1時間、インキュベートしてください。
- 速やかにセクションVIに進んでください。
VI. DNAの消化と拡散
実験開始前の準備:
!すべてのバッファーの量は、実施するCUT&RUN反応数に応じて比例的に増加させる必要があります。
Digitonin Solution #16359を90-100°Cで5分間温め、完全に解凍されて溶解していることを確認してください。解凍したDigitonin Solution #16359はすぐに氷上に置いてください。
セクションIIで固定したサンプルを用いる場合は、10% SDS Solution #20533が完全に溶解していることを確認してください。37°Cに温めることでSDSの沈殿が溶解しやすくなります。
1反応につき、Digestion Buffer (セクションVで調製したDigitonin Buffer 148.5 µL + 塩化カルシウム1.5 µL) 150 µLを調製してください。使用するまで氷上で保持してください。
1反応につき、1X Stop Buffer (CUT&RUN 4X Stop Buffer #48105 37.5 µL + Digitonin Solution #16359 3.75 µL + RNAse A (10 mg/mL) #7013 0.75 µL + Nuclease-free Water #12931 108 µL) 150 µLを調製してください。
注意:Digitonin Solution #16359は-20°Cで保管する必要があります。使用中は氷上に置き、その日の使用が終了したら-20°Cで保管してください。
- セクションVステップ5のチューブを溶液が透明になるまで (30秒間から2分間) 磁気ラックに置き、その後、液体を除去して廃棄してください。
- チューブを磁気ラックから外してください。セクションVで調製したDigitonin Buffer (+ Spermidine + PIC + Digitonin) 1 mLを加え、ピペッティングで穏やかに上下させてビーズを再懸濁してください。
- チューブを溶液が透明になるまで磁気ラックに置き (30秒間から2分間)、その後、液体を除去して廃棄してください。
- ステップ2-3をもう1度繰り返してください。
- チューブを磁気ラックから外してください。各チューブにDigestion Buffer 150 µLを加え、ピペッティングにより上下させて混合し、pAG-MNaseを活性化してください。
- サンプルを4°Cで30分間インキュベートしてください。
- 各サンプルに1X Stop Buffer (+ Digitonin + RNAse A ) を150 µL加え、ピペッティングで上下させて混合してください。
- チューブを振盪せずに37°Cで10分間インキュベートして、DNA断片を溶液中に放出させてください。
- 16,000 x gで2分間、4°Cで遠心分離して、チューブを溶液が透明になるまで磁気ラックに置いてください (30秒間から2分間)。
- 上清を新しい2 mL微量遠心分離用チューブに移してください。これが濃縮クロマチンサンプルになります。
- 固定した細胞または組織サンプルの脱クロスリンクを行うため、サンプルを室温に戻し、各サンプルに10% SDS Solution #20533 3 µL (最終濃度0.1%) およびProteinase K (20 mg/mL) #10012 2 µLを加えてください。
- 各サンプルをボルテックスで混合し、65°Cで少なくとも2時間インキュベートしてください。インキュベーションは、一晩まで延長することができます。インキュベーション後、サンプルを10,000 x gで1秒間遠心して、チューブのキャップに付着した液体を回収してください。
- サンプルの温度を室温に平衡化した後、セクションVIIIに進んでください。(SAFE STOP) ここでサンプルを-20°Cで最長1週間保存できます。ただし、DNAの精製 (セクションVIII) に移る前に、サンプルを必ず室温に戻してください。
注意:消化は4°Cの冷却ブロック上または冷蔵庫内で行ってください。氷を使用した場合は、温度が0°Cまで下がることがあり、これにより消化が制限されシグナルが低減する可能性があります。
注意:生細胞または新鮮組織 (未固定) をCUT&RUNアッセイに用いた場合、ステップ11と12を省略して、直ちにステップ13に進んでください。
注意:固定したサンプルは、後のプロトコールで65°Cでインキュベートするので、インキュベート中の蒸発を減少させるためセーフロックの付いた2 mLチューブを使用することを推奨します。
注意:サンプルが室温まで温められていない場合、SDSが沈殿する可能性があります。
VII. インプットサンプルの調製
下流解析でNGSを行う場合はインプットDNAの断片化が必要ですが、qPCRの場合は必須ではありません。ソニケーターを利用できない場合は、qPCR解析に断片化していないインプットDNAを用いることを推奨しますが、断片化していないインプットDNAはサイズが大きすぎてDNAスピンカラムで精製できないため、フェノール/クロロホルム抽出とエタノール沈殿で精製する必要があります。下流解析でNGSを行う際にソニケーターが利用できない場合は、正常IgGを用いたCUT&RUNサンプルをネガティブコントロールとして使用することもできますが、正常IgGでは非特異的なDNAが濃縮される場合があり、理想的ではありません。代替法となるMNaseを用いたインプットDNA断片化プロトコールをcst-science.com/CUT-RUN-input-digestionで公開しています。
実験開始前の準備:
! すべてのバッファーの量は、調製するインプットサンプルの数に比例して増加させる必要があります。
DNA Extraction Buffer #42015を温めてください。完全に透き通っていることを確認してください。
1インプットサンプルにつき、次の量の混合液を調製してください: Proteinase K (20 mg/mL) #10012 2 µL + RNAse A (10 mg/mL) #7013 0.5 µL + CUT&RUN DNA Extraction Buffer #42015 197.5 µL (1インプットサンプルあたり200 µL)。
- セクションII-Aのステップ7、セクションII-Bのステップ9、またはセクションII-Cのステップ16のインプットサンプル100 µLに、DNA Extraction Buffer (+ Proteinase K + RNAse A) 200 µLを加えてください。ピペッティングで上下させて混合してください。
- 最大1,200 rpmの中程度から強めで振とうしながら、チューブを55°Cで1時間インキュベートしてください。
- チューブを5分間氷上で保持し、サンプルを完全に冷却してください。
- インプットサンプルをソニケートし、細胞を溶解してクロマチンを断片化してください。ソニケーション処理の合間は、サンプルを氷上に30秒間置いてください。
- 18,500 x gで10分間、4°Cで遠心分離し、ライセートを清澄化してください。上清を新しい2 mL微量遠心分離機用チューブに移してください。
- 速やかにセクションVIII (DNAの精製) に進んでください。(SAFE STOP) ここでインプットサンプルを-20°Cで最長1週間保存できます。ただし、DNAの精製 (セクションVIII) の前には、インプットサンプルを必ず室温まで戻してください。
注意:ソニケーションの条件は、Appendix Bのプロトコールに従って、様々なソニケーターの出力設定やソニケーション処理期間を試験して決定する必要があります。100-600 bpのサイズのクロマチン断片が生成される最適なソニケーションの条件を決定してください。VirTis Virsonic 100 Ultrasonic Homogenizer/Sonicator (1/8インチプローブ) であれば、出力6に設定し、15秒パルスのソニケーションを5セット行うことで、インプットクロマチンは十分に断片化されます。
VIII. DNAの精製
DNAスピンカラム (セクションVIII-A) またはフェノール/クロロホルム抽出とエタノール沈殿 (セクションVIII-B) で、インプットサンプルや濃縮クロマチンサンプルのDNAを精製してください。DNAスピンカラムを用いた精製は簡便かつ迅速で、35 bp以上のDNA断片を効率良くに回収できます (図7A、レーン2)。フェノール/クロロホルム抽出後のエタノール沈殿による精製は比較的煩雑ですが、35 bp未満のDNA断片も回収できます (図7A、レーン3)。ただし、図7Bに示すように、CUT&RUNアッセイで得られるほとんどのDNA断片は、35 bp超です。このため、DNAスピンカラムはCUT&RUN反応による全DNA断片の>98%を精製する、迅速かつ簡便な方法と言えます。
精製したDNAは、NGS解析に進む前にPicoGreenによるDNA定量アッセイで定量できます。100,000個の細胞を用いたCUT&RUN反応で期待されるDNAの収量は、転写因子やコファクターの場合は1反応につき0.5-10 ng、ヒストン修飾の場合は1反応につき1-20 ngです。
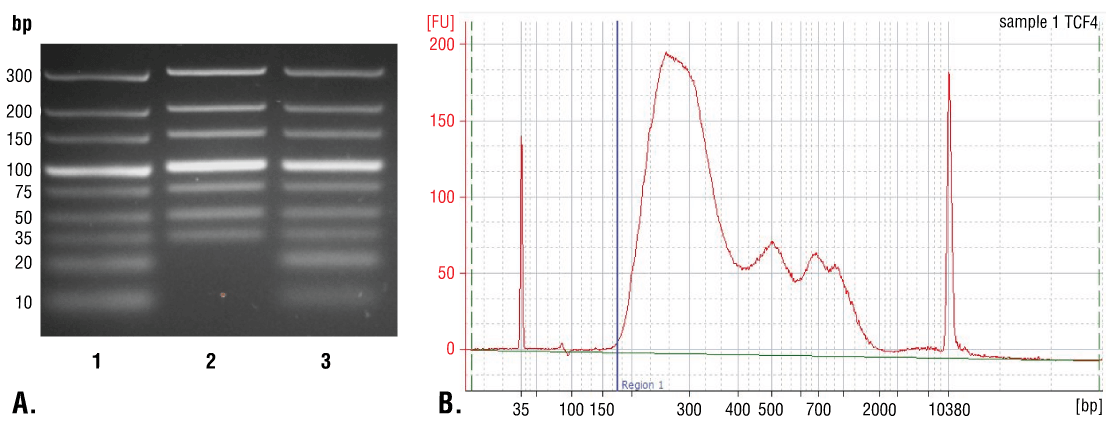
図7. DNA精製にスピンカラムを用いた場合と、フェノール/クロロホルム抽出とエタノール沈殿を用いた場合を比較しました。(A) 低分子のDNAラダーミックス (レーン1、未精製) を、DNA Purification Buffers and Spin Columns (ChIP, CUT&RUN, CUT&Tag) #14209 (レーン2) またはフェノール/クロロホルム抽出とエタノール沈殿で精製し (レーン3)、4%のアガロースゲルで電気泳動して分離しました。ここに示すように、フェノール/クロロホルム抽出とエタノール沈殿はすべてのサイズのDNA断片を効率的に回収し、DNAスピンカラムは≥35 bp以上のDNA断片を回収できます。(B) TCF4/TCF7L2 (C48H11) Rabbit Monoclonal Antibody #2569を用いたCUT&RUNアッセイで得られたDNAを、フェノール/クロロホルム抽出後のエタノール沈殿で精製しました。ライブラリーのDNA断片のサイズを、Bioanalyzer (Agilent Technologies社) を用いて解析しました。ライブラリーの構築中に付加されたアダプターやバーコード配列は、DNA断片のうちの140 bpを占めます。そのため、最初は35 bpであったDNA断片の長さは、ライブラリー調製後は175 bpになります (図中の青い縦線をご覧ください)。ここに示すように、CUT&RUNで濃縮される全DNA断片の2%未満が175 bp未満 (開始長は35 bp) であり、CUT&RUNで得られる総DNA断片の>98%をスピンカラム法で効率的に回収できることが分かります。
A. スピンカラムを用いたDNAの精製
注意:DNA Purification Buffers and Spin Columns (ChIP、CUT&RUN、CUT&Tag) #14209 (本キットには含まれません) と下記の改変プロトコールで用いることで、インプットや濃縮クロマチンサンプルからDNAを精製できます。インプットクロマチンサンプルと濃縮クロマチンサンプル 300 µLに5倍量のDNA Binding Buffer (1.5 mL) を加える要件に変更し、ステップ1から5までを改変しています。
実験開始前の準備:
!! 使用前に、DNA Wash Bufferにエタノール (96-100%) 24 mLを加えてください。このステップは、DNA精製を開始する前に1回だけ実施してください。
精製する濃縮クロマチンまたは1インプットサンプルあたり、DNA精製用コレクションチューブを1本用意します。
- 各インプットサンプルまたは濃縮クロマチンサンプルにDNA Binding Buffer 1.5 mLを加え、ピペッティングで上下させて混合してください。
- ステップ1のサンプル600 µLをそれぞれ、コレクションチューブにセットした各DNAスピンカラムに移してください。
- 18,500 x gで30秒間遠心分離してください。
- コレクションチューブから各スピンカラムを取り外し、液体を廃棄してください。空になったコレクションチューブにスピンカラムをセットしてください。
- ステップ2-4を繰り返し、ステップ1のサンプルをすべてスピンカラムに移してください。空になったコレクションチューブにスピンカラムを再度セットしてください。
- DNA Wash Buffer 750 µLをコレクションチューブにセットした各スピンカラムに加えてください。
- 18,500 x gで30秒間遠心分離してください。
- コレクションチューブから各スピンカラムを取り外し、液体を廃棄してください。空になったコレクションチューブにスピンカラムをセットしてください。
- 18,500 x gで30秒間遠心分離してください。
- コレクションチューブと液体を廃棄してください。スピンカラムは廃棄しないでください。
- 各スピンカラムにDNA Elution Buffer 50 µLを加え、清潔な1.5 mLチューブにセットしてください。
- 18,500 x gで30秒間遠心分離して、DNAを溶出してください。
- 各DNAスピンカラムを取り外し、廃棄してください。得られた溶出液が精製されたDNAです。(SAFE STOP) サンプルは、-20°Cで最長6か月間保存できます。
注意:1サンプルあたり5倍量のDNA Binding Bufferを使用します。
B. フェノール/クロロホルム抽出後にエタノール沈殿を用いたDNAの精製
注意:フェノール/クロロホルム抽出およびエタノール沈殿に必要な以下の試薬は、本キットには含まれていません:フェノール/クロロホルム/イソアミルアルコール (25:24:1)、クロロホルム/イソアミルアルコール (24:1)、3M酢酸ナトリウム (pH 5.2)、グリコーゲン 20 mg/mL、100%エタノール、70%エタノールおよび1X TEバッファーまたはNuclease-free Water #12931。
- フェノール/クロロホルム/イソアミルアルコール (25:24:1) 300 µLをインプットサンプルまたは濃縮クロマチンサンプルに加え、30秒間ボルテックスして十分に混合してください。
- 16,000 x gで5分間遠心分離して、層状に分離させてください。上部の水層の大部分を (中間層を避けて) 慎重に新しいチューブに移してください。
- クロロホルム/イソアミルアルコール (24:1) 300 µLを水層サンプルに加え、30秒間ボルテックスして十分に混合してください。
- 16,000 x gで5分間遠心分離して、層状に分離させてください。上部の水層の大部分を (中間層を避けて) 慎重に新しいチューブに移してください。
- 3M酢酸ナトリウム (pH 5.2) 25 µL、20 mg/mLグリコーゲン 1 µL、100%エタノール 300 µLを各水層サンプルに加え、30秒間ボルテックスして混合してください。
- -80°Cに1時間、あるいは-20°Cに一晩置いて、DNAを沈殿させてください。
- 4°C、16,000 x gで5分間遠心分離して、DNAをペレットにしてください。
- 上清を慎重に除去して廃棄し、70%エタノールでペレットを洗浄してください。
- 4°C、16,000 x gで5分間遠心分離して、DNAをペレットにしてください。
- 上清を除去し、ペレットを風乾してください。
- 1X TEバッファーまたはNuclease-free Water #12931 50 µLにペレットを再懸濁してください。これが精製されたDNAです。(SAFE STOP) サンプルは、-20°Cで最長6か月間保存できます。
IX. qPCRによるDNA定量
推奨事項:
本キットに含まれるDrosophila Normalization Primer Setは、Drosophila melanogasterのCG3402遺伝子に特異的であり、Drosophila Spike-In Nuclei Controlのシグナルの定量によるサンプルの正規化に用います。
キットにはコントロールプライマーとしてヒトあるいはマウスのRPL30遺伝子 (#7014あるいは#7015) が含まれており、Tri-Methyl-Histone H3 (Lys4) (C42D8) Rabbit Monoclonal Antibody #9751サンプルの定量的リアルタイムPCRに使用できます。他の生物種でCUT&RUNを実施する場合は、その生物種に適切なコントロールプライマーを設計し、最適なPCR条件を決定してください。
プライマーの設定は非常に重要です。CUT&RUNの場合、PCR増幅産物のサイズは約60-80 bpとなるのが望ましいです。プライマーをデザインする場合、融解温度は約60°C、GC含量は約50%が最適です。
qPCRでヒストンや転写因子、コファクターの標的遺伝子を定量する場合、qPCRに用いる精製DNAは2 µLで十分です。
非特異的なPCR産物の増幅を防ぐため、ホットスタート用Taqポリメラーゼの使用を推奨します。
コンタミネーションを防ぐため、フィルターチップ付きピペットを使用してください。
- 使用するPCR装置のモデルに対応するPCRチューブあるいはPCRプレートに、正しくサンプル番号を記載してください。PCR反応には、ポジティブコントロールのtri-methyl-histone H3 Lys4サンプル、ネガティブコントロールのRabbit IgGサンプル、DNAコンタミネーションに対するコントロールとしてDNAなしのチューブ、インプットDNAサンプルを含めてください。必要であれば、インプットDNAの段階希釈 (未希釈-100%インプット、1:5-20%インプット、1:25-4%インプット、1:125-0.8%インプット) を用いて標準曲線を作成して増幅効率を決定し、各免疫濃縮サンプル内のDNA量を定量してください。
- 各PCRチューブまたはPCRプレートの各ウェルに、DNAサンプル 2 µLを分注してください。
- 下記の通り、マスターミックスを調製してください。各PCR反応に対し、2-3個の複製サンプルを用意してください。分量の損失を考慮して、十分量のマスターミックスを調製してください (1-2反応分を余計に調製するなど)。PCRチューブまたは各ウェルにマスターミックス18 µLを加えてください。
- PCR反応を以下のプログラムで開始してください:
- リアルタイムPCR装置に付属のソフトウェアを使用して、定量結果を解析してください。代替法として、Percent Input法により下記の公式を用いて、免疫沈降の効率を算出することもできます。この方法では、各抗体反応で回収されたシグナルを、インプットクロマチン総量に対する割合 (%) で算出します。インプットDNAサンプルの段階希釈を使用する場合、% Input (100%、20%、4%、0.8%) のLog(10) に対する標準曲線をプロットし、使用して各抗体反応で得られたシグナルを算出します。
- Percent Input = 100% x 2(C[T] 100% Input Sample - C[T] IP Sample)
- C[T] = Ct = Cq = PCR反応の平均閾値サイクル
- サンプルを正規化する際は、Drosophila Normalization Primer SetのC[T]値が最も低いサンプルを基準サンプルとして選択してください (例:下表のサンプル1)。次の公式を用いて、その他のサンプルすべての正規化係数を算出してください。算出した正規化係数を用いて、各プライマーセットのシグナルを調整してください。
注意:CUT&RUNサンプルのみをDrosophila Normalization Primer Setを用いて解析します。インプットDNAには、qPCRテンプレートとして使用するDrosophila Spike-In Nuclei Controlが含まれていません。
|
試薬 |
PCR反応1回の分量 (18 µL) |
|---|---|
| Nuclease-free Water #12931 | 6 µL |
| 5 µMのプライマー | 2 µL |
| SimpleChIP® Universal qPCR Master Mix #88989 | 10 µL |
| a. | 初期変性 | 95°C、3分間 |
| b. | 変性 | 95°C、15秒間 |
| c. | アニーリングおよび伸長 | 60°C、60秒間 |
| d. | 合計40サイクルとなるようにステップb-cを繰り返してください。 |
Tri-Methyl-Histone H3 (Lys4) (C42D8) Rabbit Monoclonal Antibody #9751 and SimpleChIP® Human β-Actin Promoter Primers #13653を用いたqPCRアッセイのサンプル標準化の例 (図8を参照してください)
| Dm Normalization Primer SetのC[T] 値 | **qPCRの正規化係数 | 正規化前のシグナル (%インプット) | 正規化後のシグナル (%インプット) | |
| サンプル1 | 26.72 | 2(26.72-26.72)=1.00 | 0.99% | 0.99%/1.00=0.99% |
| サンプル2 | 27.61 | 2(26.72-27.61)=0.54 | 0.61% | 0.61%/0.54=1.13% |
| サンプル3 | 29.29 | 2(26.72-29.29)=0.17 | 0.20% | 0.20%/0.17=1.18% |
| サンプル4 | 31.08 | 2(26.72-31.08)=0.05 | 0.06% | 0.06%/0.05=1.20% |
**qPCRの正規化係数 = 2 (C[T] 基準サンプル – C[T] その他のサンプル)
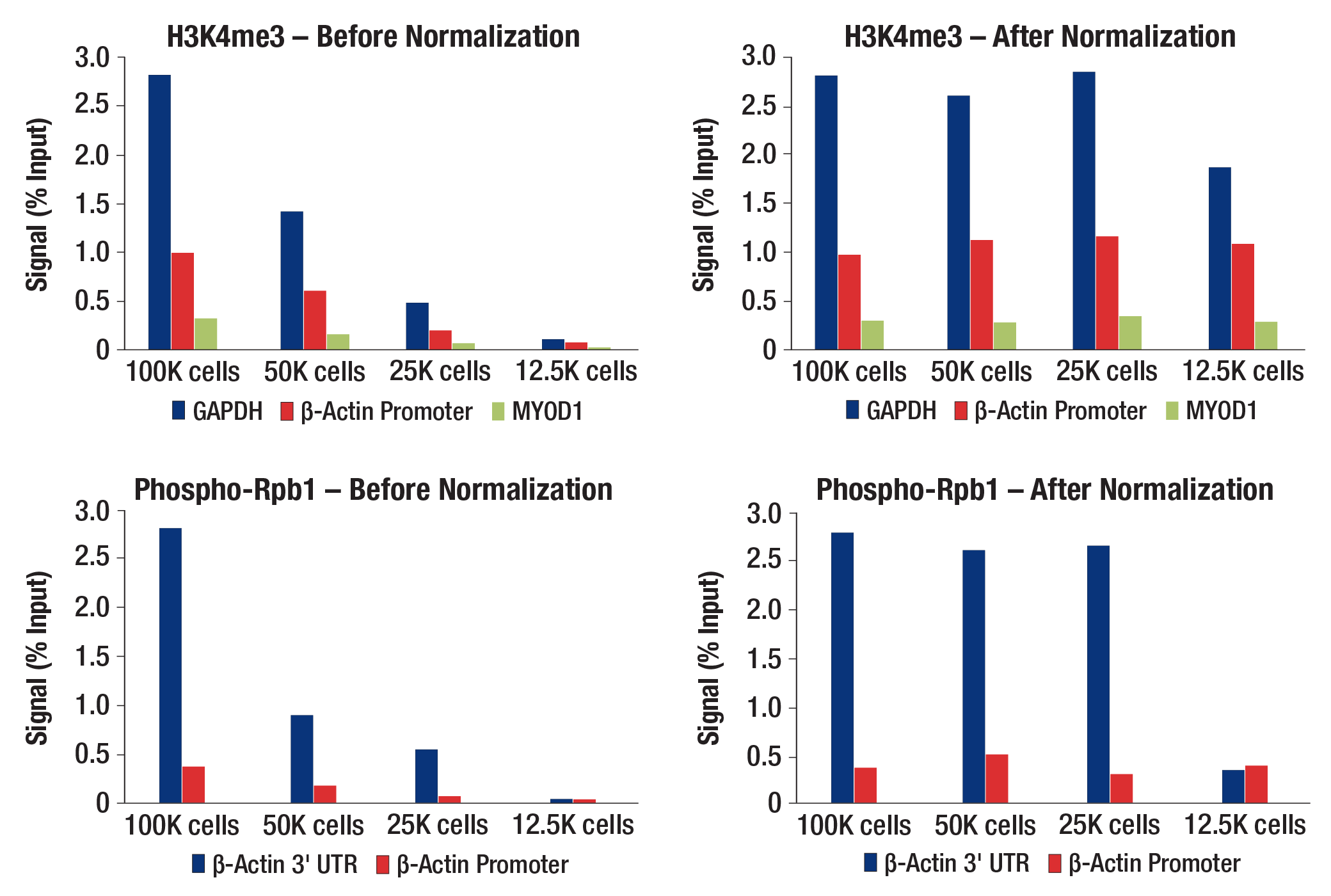
図8. qPCR解析におけるDrosophila Spike-In Nuclei Controlを用いたCUT&RUNシグナルの正規化。異なる細胞数のHCT116細胞に対して、Tri-Methyl-Histone H3 (Lys4) (C42D8) Rabbit Monoclonal Antibody #9751 (上のパネル) あるいはPhospho-Rpb1 CTD (Ser2) (E1Z3G) Rabbit Monoclonal Antibody #13499 (下のパネル) を用いてCUT&RUNを実施しました。濃縮DNAを、human GAPDH exon 1 primers、SimpleChIP® Human beta-Actin Promoter Primers #13653、SimpleChIP® Human beta-Actin 3' UTR Primers #13669、SimpleChIP® Human MyoD1 Exon 1 Primers #4490を用いて、リアルタイムPCRで解析しました。CUT&RUNで回収されたDNA量を、Inputクロマチンに対する相対量 (100,000細胞の総クロマチン量を100%とした相対値) で示しています。また、正規化前の濃縮の結果を左のパネルに示しています。最初の細胞数に比例させた量のDrosophila Spike-In Nuclei Controlを、各反応に加えました。各サンプル中のDrosophila Normalization Primer Setを用いて取得したDrosophila Spike-In Nuclei ControlからのqPCRシグナルの差に基づいて、各サンプルのCUT&RUNシグナルを100,000個の細胞分になるように正規化しました。標準化後の濃縮結果を右のパネルに示しています。サンプルのインプット量が極めて少ないためにCUT&RUNアッセイが失敗した場合でも、正規化後のシグナルはアーティファクトな高シグナルを生成することなく、失敗という結果を正確に反映する点は重要です。これにより、この正規化戦略の信頼性と正確性が実証されます (12.5Kの細胞を用いたRpb1データを参照してください)。
X. NGSライブラリーの構築
CUT&RUNキットを用いて調製したDNAサンプルは、直接NGSに使用できます。NGS DNAライブラリーの構築の際は、使用予定のシーケンスプラットフォームと互換性のあるDNAライブラリー調製プロトコールまたはキットを使用してください。Illuminaシステムプラットフォームによるシーケンシングの場合は、DNA Library Prep Kit for Illumina Systems (ChIP-seq, CUT&RUN) #56795を、Multiplex Oligos for Illumina Systems (ChIP-seq, CUT&RUN) #29580または#47538と共にCUT&RUN DNAのプロトコールに従って使用することを推奨します。
CUT&RUNのバックグラウンドシグナルは非常に低いため、通常はヒストン修飾や転写因子の解析のシーケンシング深度は、1サンプルあたり500万リードで十分です。シーケンシング深度が1サンプルあたり1,500万以上の場合、リードの重複率が大幅に上昇します。シーケンシング深度が1サンプルあたり200万以下の場合、S/N比が低下します。
使用した細胞数が20,000個未満の場合は、一般にNGSで得られるリードのマッピング率が低下し、重複率が上昇します。このような場合、下流のデータ解析に十分なユニークマップされたリード数を得るため、シーケンシング深度を上げることを推奨します。
サンプルを正規化する際は、全サンプルからのCUT&RUNシーケンシングデータを、試験用ゲノム (例えば、ヒト、マウス、その他) およびショウジョウバエゲノムの両方にマッピングしてください。各サンプルについて、全ユニークリード数に対するSpike-inリード数の比率を算出してください。比率が最も低いサンプルをリファレンスサンプルとして選択し (例:下表のサンプル4)、得られた式を用いてその他のサンプルの正規化係数を算出してください。バイオインフォマティクス解析において、各サンプルのbigWigファイルをスケーリング (例:deepToolsのbamCoverageを使用) するか、あるいは試験ゲノムにアライメントされたユニークリード数をダウンサンプリングしてこれらの係数を適用してください。シーケンシング深度がサンプル間で一貫している場合、正規化係数はSpike-inリード数と全リード数の比率ではなく、Spike-inリードの絶対数を用いて算出することもできます。
細胞のタイトレーション実験におけるTri-Methyl-Histone H3 (Lys27) (C36B11) Rabbit Monoclonal Antibody #9733を用いたNGSアッセイサンプルmp正規化の例 (図2を参照してください)
| Spike-inゲノムにアラインメントされたユニークリード数 (dm6) | 解析対象のリファレンスゲノムにアラインメントしたユニークリード数 (hg38) | 全ユニークリード数に対するユニークSpike-inリード数の比率 | NGSの正規化係数 | |
| サンプル1 | 262,459 | 1,160,970 | 262,459/(262,459 + 1,160,970) = 0.184 | 0.034/0.184 = 0.19 |
| サンプル2 | 113,312 | 1,027,248 | 113,312/(113,312 + 1,027,248) = 0.099 | 0.034/0.099= 0.34 |
| サンプル3 | 184,441 | 2,998,350 | 184,441/(184,441 + 2,998,350) = 0.058 | 0.034/0.058= 0.59 |
| サンプル4 | 66,880 | 1,901,014 | 66,880/(66,880 + 1,901,014) = 0.034 | 0.034/0.034= 1 |
NGSの正規化係数 = コントロールサンプルにおけるユニークSpike-inリード数と全ユニークリード数との比率/その他のサンプルにおけるユニークSpike-inリード数と全ユニークリード数との比率
APPENDIX A:細胞のジギトニンに対する感受性の決定
CUT&RUNのプロトコールでは、バッファーにジギトニンを添加することで細胞膜の透過化を行い、一次抗体やpAG-MNase酵素が細胞や核に進入できるようにします。このため、抗体と酵素の結合および標的ゲノム遺伝子座の消化には、適量のジギトニンを含むバッファーが不可欠です。各細胞株は、ジギトニンの細胞透過化に対して異なる感受性を示します。本プロトコールで推奨されているジギトニンの量は、ほとんどの細胞株や組織の透過化に十分ですが、下記のプロトコールを用いて、使用する特定の細胞株や組織のジギトニン感受性試験を行うこともできます。過剰なジギトニンの添加はアッセイに対して有害ではないことが分かっているため、濃度曲線を作成する必要はありません。推奨されるジギトニンの量が使用する細胞株に十分かどうかは、簡単な試験により判断できます。
実験開始前の準備:
Digitonin Solution #16359を90-100°Cで5分間温め、完全に解凍されて溶解していることを確認してください。解凍したDigitonin Solution #16359はすぐに氷上に置いてください。
細胞または組織サンプルにつき、Wash Buffer 100 µL (10X Wash Buffer (CUT&RUN、CUT&Tag) #31415 10 µL + Nuclease-free Water #12931 90 µL) を調製してください。この試験には、100X Spermidine #27287やProtease Inhibitor Cocktail (200X) #7012を加える必要はありません。
注意:Digitonin Solution #16359は-20°Cで保管する必要があります。使用中は氷上に置き、その日の使用が終了したら-20°Cで保管してください。
- 1.5 mLチューブに10,000-100,000細胞を回収してください。組織の場合は、1 mgの組織から乖離した細胞を回収します (セクションII-C、ステップ1-15)。
- 600 x gで3分間、室温で遠心分離し、液体を除去して廃棄してください。
- Wash Buffer 100 µLに細胞ペレットを再懸濁してください。
- 1反応あたり、Digitonin Solution #16359を2.5 µL加え、室温で10分間インキュベートしてください。
- 細胞懸濁液10 µLと0.4% トリパンブルー色素 10 µLを混合してください。
- 血球計算盤またはセルカウンターを使用して、染色された細胞数と総細胞数を計測してください。透過化が十分であれば、>90%の細胞がTrypan Blueで染色されます。
- 90%未満の細胞しかトリパンブルーで染色されない場合は、ジギトニンバッファーに加えるDigitonin Solution #16359の量を増加させて、>90%の細胞が透過化され染色されるまで、ステップ1-6を繰り返してください。セクションIV-VIでは、この量のDigitonin Solution #16359を使用してください。
注意:細胞ペレットが肉眼で確認できない場合は、ステップ2で細胞懸濁液を最初に遠心分離した後、ペレットを崩さない程度になるべく多くの培地を除去し、1反応あたり細胞培地を≤40 µL残すことを推奨します。続くステップ3で、適量の1X Wash Bufferを加え、細胞懸濁液の総量を100 µLにしてください。
APPENDIX B:インプットサンプルのソニケーションの最適化
DNAスピンカラムを使用して精製できるのは、断片化されたゲノムDNA (<10 kb) だけであることから、インプットDNAサンプルのソニケーションを推奨します。断片化されたゲノムDNA (<1 kb) は、NGS解析でネガティブコントロールとして使用できます。インプットDNAの長さが100-600 bpになるように、ソニケーションを最適化する必要があります。
NGSのコントロールには、インプットサンプルを使用することを推奨します。インプットサンプルは、偏りのない細胞ゲノムの代表として簡便に使用できるからです。IgGサンプルもNGSのネガティブコントロールとして使用できますが、非特異的な結合によりゲノムの特定領域で濃縮が見られる場合があります。qPCR解析の場合は、インプットDNAを断片化せずに使用することができます。ただし、断片化されていないDNAは、フェノール/クロロホルム抽出とエタノール沈殿で精製する必要があります。
実験開始前の準備:
! すべてのバッファーの量は、調製するインプットサンプルの数に比例して増加させる必要があります。
CUT&RUN DNA Extraction Buffer #42015を室温に戻し、完全に解凍されて液状になっていることを確認してください。
1インプットサンプルにつき、1X Wash Buffer (10X Wash Buffer (CUT&RUN, CUT&Tag) #31415 210 µL + Nuclease-free Water #12931 1.89 mL) 2.1 mLを調製してください。細胞へのストレスを最小限にするため、室温に戻してください。この試験には、100X Spermidine #27287やProtease Inhibitor Cocktail (200X) #7012を加える必要はありません。
1インプットサンプルにつき、次の量の混合液を調製してください:Proteinase K #10012 2 µL + RNAse A (10 mg/mL) #7013 0.5 µLを加え、CUT&RUN DNA Extraction Buffer #42015 197.5 µLを調整してください (インプット1サンプルあたり200 µL)。
- 1.5 mLチューブ中に、検討する各ソニケーション条件につき、CUT&RUN実験で使用するインプットと同じ数の細胞 (5,000-100,000細胞) を回収してください。組織の場合、各ソニケーション条件につき、CUT&RUN実験で使用するインプットと同じ量の組織から解離させた細胞を回収してください (セクションII-C ステップ1-15)。
- 600 x gで3分間、室温で遠心分離し、液体を除去して廃棄してください。
- 1X Wash Buffer 1 mLを加えて、ピペッティングで穏やかに上下させて細胞ペレットを再懸濁してください。
- 600 x gで3分間、室温で遠心分離し、液体を除去して廃棄してください。
- ステップ3-4を繰り返して、もう1度細胞ペレットを洗浄してください。
- 各ソニケーション条件あたり、1X Wash Buffer 100 µLを加え、穏やかにピペッティングで上下させて細胞ペレットを再懸濁してください。
- 細胞懸濁液100 µLを、各ソニケーション条件ごとに新しいチューブに分注してください。
- 各サンプルにCUT&RUN DNA Extraction Buffer (+ Proteinase K + RNAse A) 200 µLを加えて、ピペッティングで上下させて混合してください。
- 最大1,200 rpmの中程度から強めで振とうしながら、チューブを55°Cで1時間インキュベートしてください。
- チューブを5分間氷上で保持し、サンプルを完全に冷却してください。
- お使いのソニケーターでの最適なソニケーション条件を決定するために、15秒間のパルスソニケーションのサイクル数を増加させながら、タイムコース実験を実施してください。ソニケーション処理の合間は、サンプルを氷上に30秒間置いてください。
- 18,500 x gで10分間、4°Cで微量遠心分離機を用いて遠心分離して、ライセートを清澄化してください。上清を新しい2 mL微量遠心分離用チューブに移してください。
- セクションVIIIに従って、DNAスピンカラム法、またはフェノール/クロロホルム抽出後のエタノール沈殿により、DNAサンプルを精製してください。
- カラムからDNAを溶出するか、DNAペレットを1X TEバッファーまたはNuclease-free Water #12931 30 µLで再懸濁してください。
- 電気泳動でDNA断片のサイズを決定してください。100 bp DNAマーカーと共に>15 µLのサンプルを、1%アガロースゲルにロードしてください。ゲル上のDNAスメアを観察するため、色素フリーのローディングバッファー (30%グリセロール) の使用を推奨します。
- 最適なサイズである100-600 bpのDNA断片が得られるソニケーション条件を決定し、セクションVIIステップ4のインプットサンプルの調製を実施してください。最適なソニケーション条件が得られない場合は、ソニケーターの出力設定あるいはソニケーションのサイクル数を増減させて、ソニケーションのタイムコース実験を繰り返してください。
注意:使用細胞数が少なく (<100,000細胞)、遠心分離で集めた細胞ペレットが肉眼で確認できない場合は、下記ステップ3-5の洗浄操作を省略することを推奨します。ステップ2で細胞懸濁液を最初に遠心分離した後、ペレットを崩さない程度になるべく多くの培地を除去し、細胞培地を≤40 µL残すことを推奨します。この場合、ステップ6で細胞懸濁液に適量の1X Wash Bufferを加え、各ソニケーション条件あたりの合計容量を100 µLにしてください。
注意:このサンプルは、ステップ9で55°Cでインキュベートするため、インキュベート中の蒸発を低減させるためにセーフロックの付いた1.5 mLチューブを使用することを推奨します。
APPENDIX C:トラブルシューティングガイド
詳細なトラブルシューティングガイドについては、//cst-science.com/troubleshooting-CUT+-RUN をご覧ください。
APPENDIX D:オンラインリソース
CUT&RUNアッセイに関するプロトコールビデオやセミナー、ポスター、ブログ、よくある質問、詳細情報についてはCUT&RUNリソースセンター (cellsignal.com/applications/cut-and-run) にアクセスするか、以下のQRコードをスキャンしてご覧ください。
